
The Protein Structure Prediction
Lindert Lab Research
Computational Protein Structure Prediction
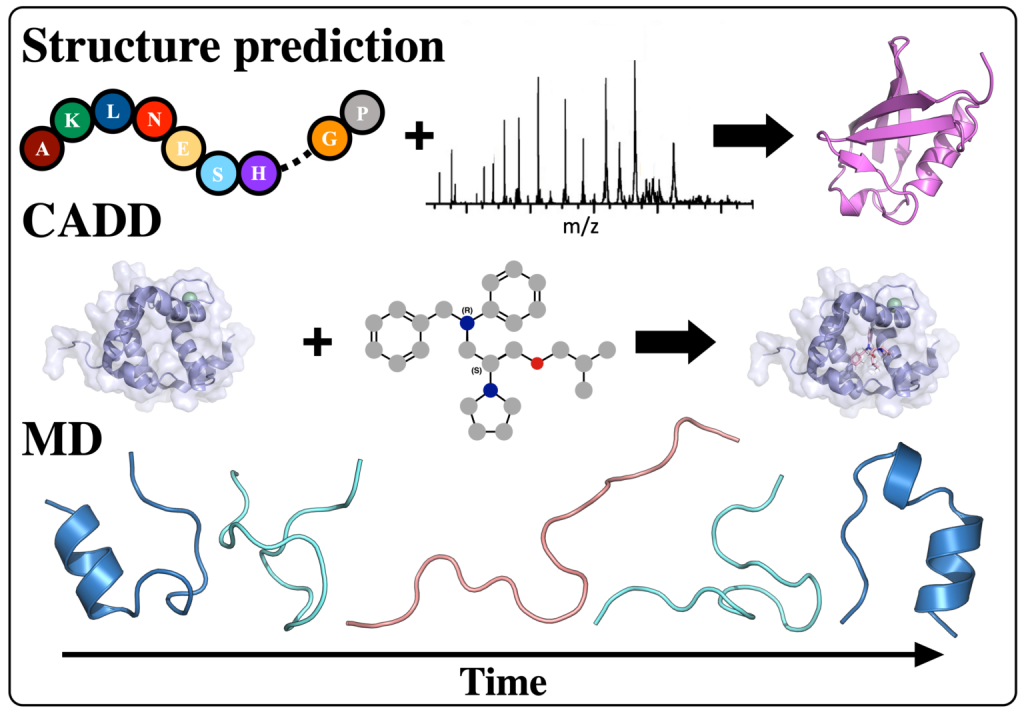
Protein structure prediction is a highly successful computational method, offering a promising solution in cases where experimental techniques fail to determine protein structure. By combining sparse experimental data with computational modeling techniques, our lab has been able to make a notable contribution to this field. De novo structure prediction has been most successful when combined with sparse experimental data, forming a symbiosis where the combination of methods is more powerful than the sum of the individuals. The Lindert Lab is an active member of the Rosetta Commons Community, and we work to develop new algorithms for protein structure prediction which incorporate sparse experimental data from a variety of sources such as mass spectrometry, cryo-EM, and NMR. In recent years, artificial intelligence has made tremendous strides in protein structure prediction, and the lab is interested in incorporating machine learning/deep learning methods into all aspects of our current structure prediction projects.
Protein Structure Prediction Reviews
- Biehn, S.E.; Lindert, S. (2021), Protein Structure Prediction with Mass Spectrometry Data. Annu Rev Phys Chem. 73, 1-19. Seffernick, J.T.; Lindert, S. (2020), Hybrid methods for combined experimental and computational determination of protein structure. J Chem Phys 153 (24), 240901.
- Lindert, S. (2021), Protein Structure Prediction with Mass Spectrometry Data. Annu Rev Phys Chem. 73, 1-19.
Computational Protein Structure Prediction
Protein structure prediction is a highly successful computational method, offering a promising solution in cases where experimental techniques fail to determine protein structure. By combining sparse experimental data with computational modeling techniques, our lab has been able to make a notable contribution to this field. De novo structure prediction has been most successful when combined with sparse experimental data, forming a symbiosis where the combination of methods is more powerful than the sum of the individuals. The Lindert Lab is an active member of the Rosetta Commons Community, and we work to develop new algorithms for protein structure prediction which incorporate sparse experimental data from a variety of sources such as mass spectrometry, cryo-EM, and NMR. In recent years, artificial intelligence has made tremendous strides in protein structure prediction, and the lab is interested in incorporating machine learning/deep learning methods into all aspects of our current structure prediction projects.
Protein Structure Prediction Reviews
- Biehn, S.E.; Lindert, S. (2021), Protein Structure Prediction with Mass Spectrometry Data. Annu Rev Phys Chem. 73, 1-19. Seffernick, J.T.; Lindert, S. (2020), Hybrid methods for combined experimental and computational determination of protein structure. J Chem Phys 153 (24), 240901.
- Lindert, S. (2021), Protein Structure Prediction with Mass Spectrometry Data. Annu Rev Phys Chem. 73, 1-19.
Cryogenic Electron Microscopy (cryo-EM)
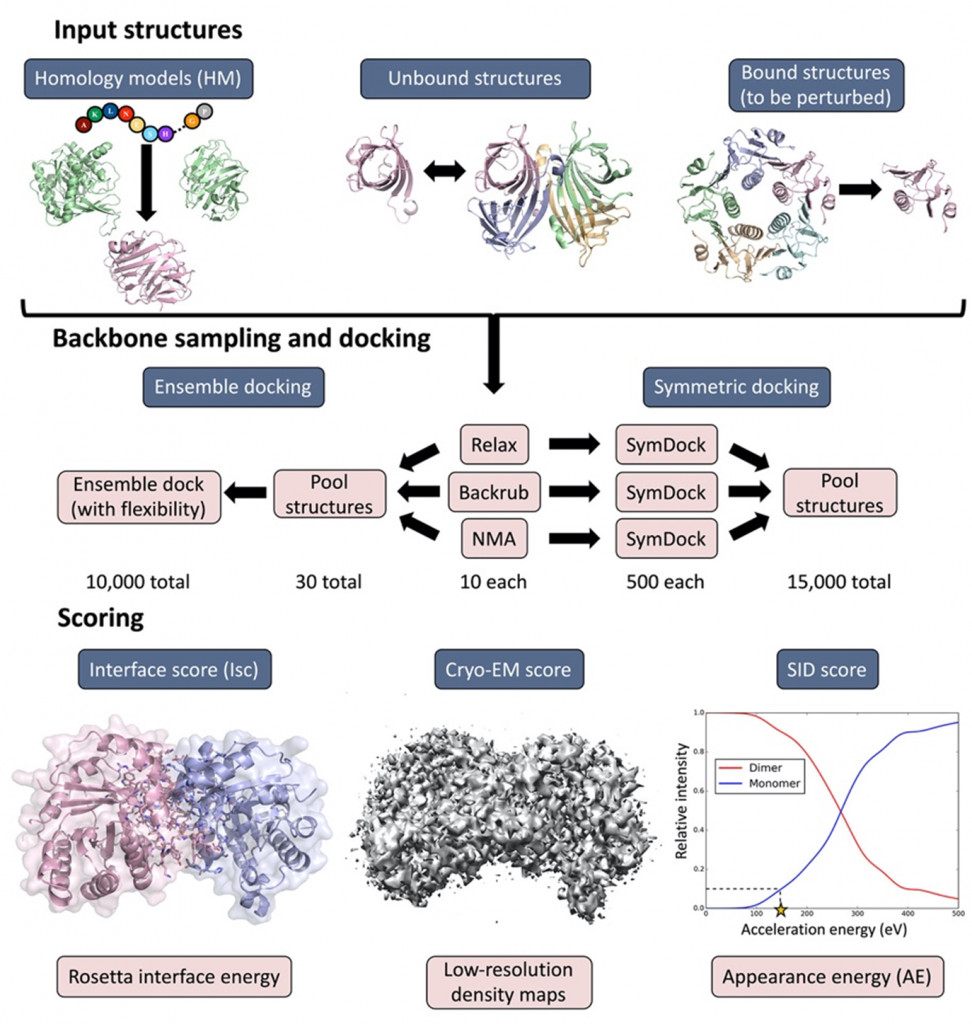
Cryo-EM has become a primary method for protein structure elucidation, frequently yielding density maps with near-atomic or medium resolution. If protein structures cannot be determined unambiguously from the density maps, computational structure refinement tools are needed to generate protein structural models. We have demonstrated that using Rosetta and Molecular Dynamics guided by medium-resolution cryo-EM density maps can lead to highly accurate protein structure predictions. We continue to use medium to low resolution cryo-EM density maps in combination with other structural techniques, such as surface-induced dissociation, to improve protein structure elucidation.
Related Reviews
- Seffernick, J.T.; Canfield, S.M.; Harvey, S.R.; Wysocki, V.H.; Lindert, S. (2021), Prediction of Protein Complex Structure Using Surface-Induced Dissociation and Cryo-Electron Microscopy. Anal Chem 93 (21), 7596–7605.
- Lindert, S. (2021), Protein Structure Prediction with Mass Spectrometry Data. Annu Rev Phys Chem. 73, 1-19.
Nuclear Magnetic Resonance (NMR)
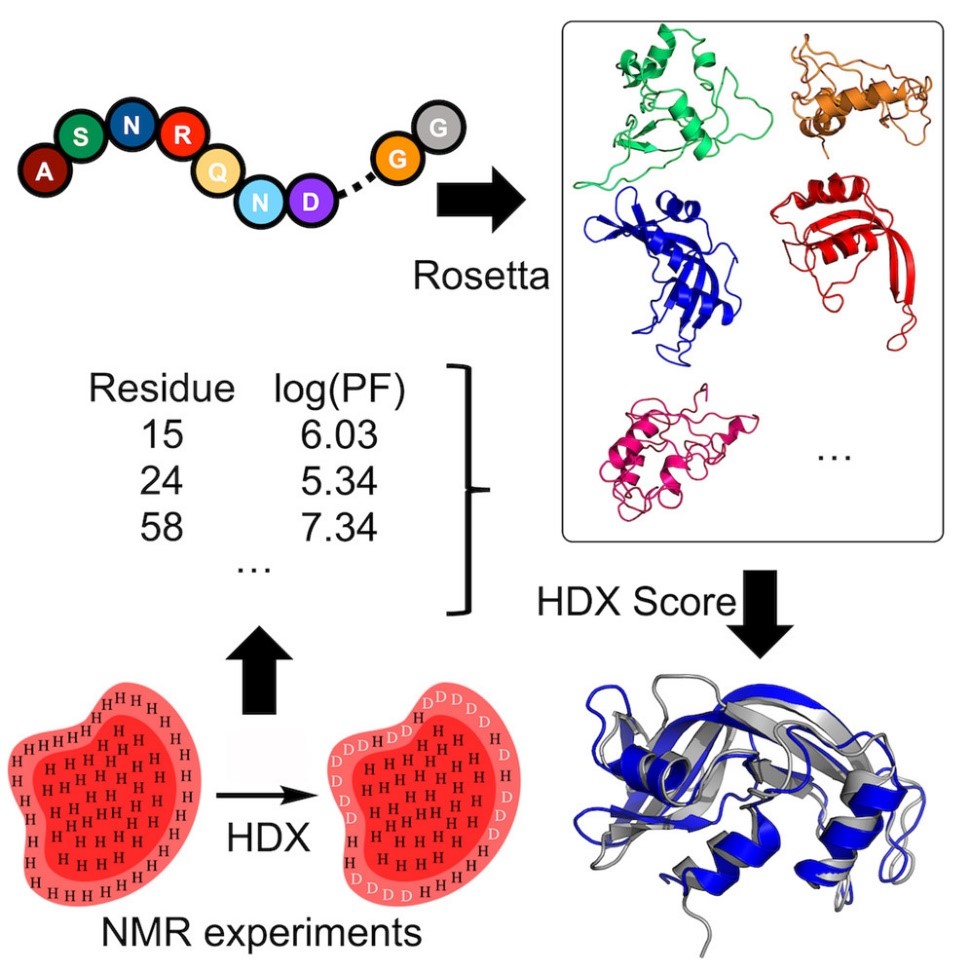
Hydrogen-deuterium exchange (HDX) measured by NMR provides protein structural information relating to residue solvent accessibility and flexibility. While this structural information is beneficial, it is insufficient to elucidate structures unambigiously. Thus, computational methods are needed to translate the HDX-NMR data into protein structures. We have developed a methodology to incorporate HDX-NMR data (categories of exchange rate and protection factors) into ab initio protein structure prediction using the Rosetta software to predict structures based on experimental agreement.
Related Publications
-
Nguyen, T.T.; Marzolf, D.R.; Seffernick, J.T.; Heinze, S.; Lindert, S. (2021), Protein structure prediction using residue-resolved protection factors from hydrogen-deuterium exchange NMR. Structure 30 (2), 313-320.
- Marzolf, D.R.; Seffernick, J.T.; Lindert, S. (2021), Protein Structure Prediction from NMR Hydrogen-Deuterium Exchange Data. J Chem Theory Comput 17 (4), 2619-2629. Leelananda, S.P.; Lindert, S. (2020), Using NMR Chemical Shifts and Cryo-EM Density Restraints in Iterative Rosetta-MD Protein Structure Refinement. J Chem Inf Model 60 (5), 2522-2532.
Simulation of Biomolecules
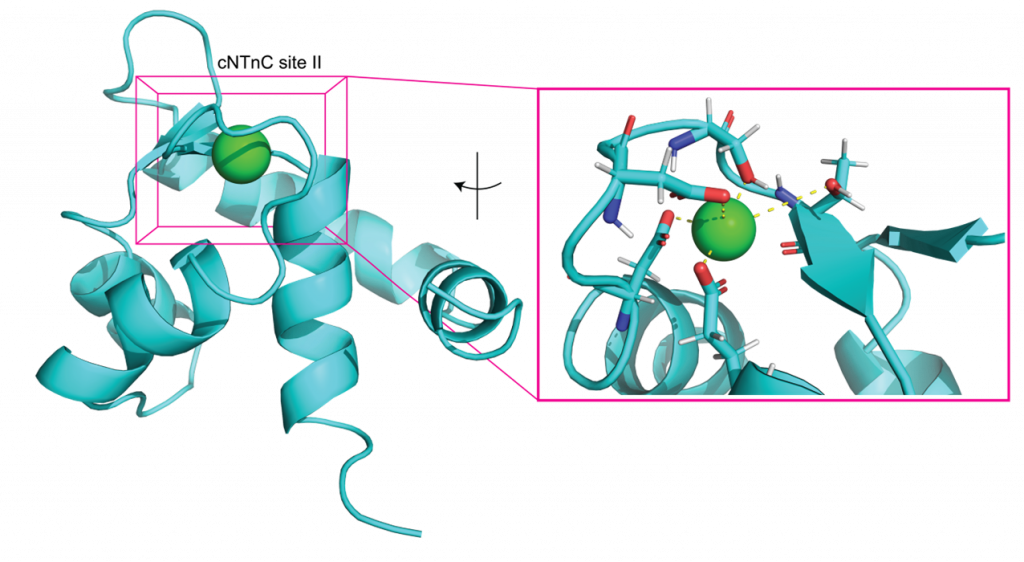
The Lindert lab is interested in understanding the relationship between dynamics, structure and function in proteins. For many biological systems the dynamics are crucial to their function. We use Molecular Dynamics (MD) simulations to investigate transitions between different conformational states, elucidate the dynamics of binding pockets (with implications for drug discovery) and gain insight into free energy barriers between states. Enhanced sampling methods, such as Gaussian accelerated MD (GaMD) can provide a means of extending the sampling of the conformational space beyond the current limits of nanosecond to microsecond timescales. Additionally, the lab makes use of various free energy methods such as umbrella sampling, thermodynamic integration, and adaptive steered molecular dynamics to quantitatively assess transitions between different thermodynamic states. The lab’s focus is on simulations of contraction in muscle cells. Both in cardiac and skeletal muscle, thin and thick filaments slide past each other to achieve muscle contraction. The process of contraction is regulated by calcium and encompasses multiple molecular events. Thus, our simulations target different proteins involved in contraction and also look at different scales (from a single protein to macromolecular complexes), with a particular focus on MD simulations of the cardiac troponin complex. The results of these simulations are then used to aid the identification of drugs targeting heart failure.L
Related Publications
- Troponin Reviews: Bowman, J.D.; Lindert, S. (2019), Computational Studies of Cardiac and Skeletal Troponin. Front Mol Biosci 6:68
- Hantz, E.R.; Lindert, S. (2022), Computational Exploration and Characterization of Potential Calcium Sensitizing Mutations in Cardiac Troponin C. J Chem Inf Model 62 (23), 6201-6208. Hantz, E.R.; Lindert, S. (2021), Adaptive Steered Molecular Dynamics Study of Mutagenesis Effects on Calcium Affinity in the Regulatory Domain of Cardiac Troponin C. J Chem Inf Model 61 (6), 3052-3057.
- Hantz, E.R.; Lindert, S. (2021), Adaptive Steered Molecular Dynamics Study of Mutagenesis Effects on Calcium Affinity in the Regulatory Domain of Cardiac Troponin C. J Chem Inf Model 61 (6), 3052-3057. Rayani, K.; Seffernick, J.T.; Li, A.Y.; Davis, J.P.; Spuches, A.M.; Van Petegem, F.; Solaro, R.J.; Lindert, S.; Tibbits, G.F. (2021), Binding of calcium and magnesium to human cardiac Troponin C. J Biol Chem 296, 100350.
Sponsors & Partners
Our Research Partners

Newsletter
Lorem ipsum dolor sit amet, consectetur adipiscing elit. Ut elit tellus, luctus nec ullamcorper mattis, pulvinar dapibus leo.